
In the first part of this blog series, I provided an overview of the ProteomeXChange (PX) Consortium and introduced its most important member – the PRIDE Database. I also introduced ProteomeCentral, the common portal providing the ability to browse and search all of the public datasets within the PX Consortium member repositories. Here in the second part of this blog series, I will cover the remaining five members of the PX Consortium.
- Mass Spectrometry Interactive Virtual Environment (MassIVE)
MassIVE is a community resource developed by the NIH-funded Center for Computational Mass Spectrometry at the University of California, San Diego to promote the global, free exchange of mass spectrometry data. MassIVE is designed not only for reliable sharing of primary data but also offers user-friendly tools for advanced data analysis and visualization.
Datasets that are directly submitted to MassIVE make up about 6% of all datasets in ProteomeCentral. MassIVE was developed along with the Global Natural Product Social Molecular Networking (GNPS) ecosystem. As the result, it also plays a significant role in metabolomics. More importantly, MassIVE systematically replicates datasets from other PX repositories as well as from the now extinct Tranche repository. This is a major effort from MassIVE to fulfill its mission as a community resource to promote the global, free exchange of MS data.
Currently, MassIVE contains over 15,000 public datasets, making it the second largest proteomics repository after PRIDE. Datasets in MassIVE are assigned unique IDs (“MSV” followed by nine digits). In addition, datasets that are also submitted to ProteomeCentral are assigned with PX Accession Numbers (“PXD” followed by six digits). However, not all proteomics datasets in MassIVE are included in ProteomeCentral (Figure 1).
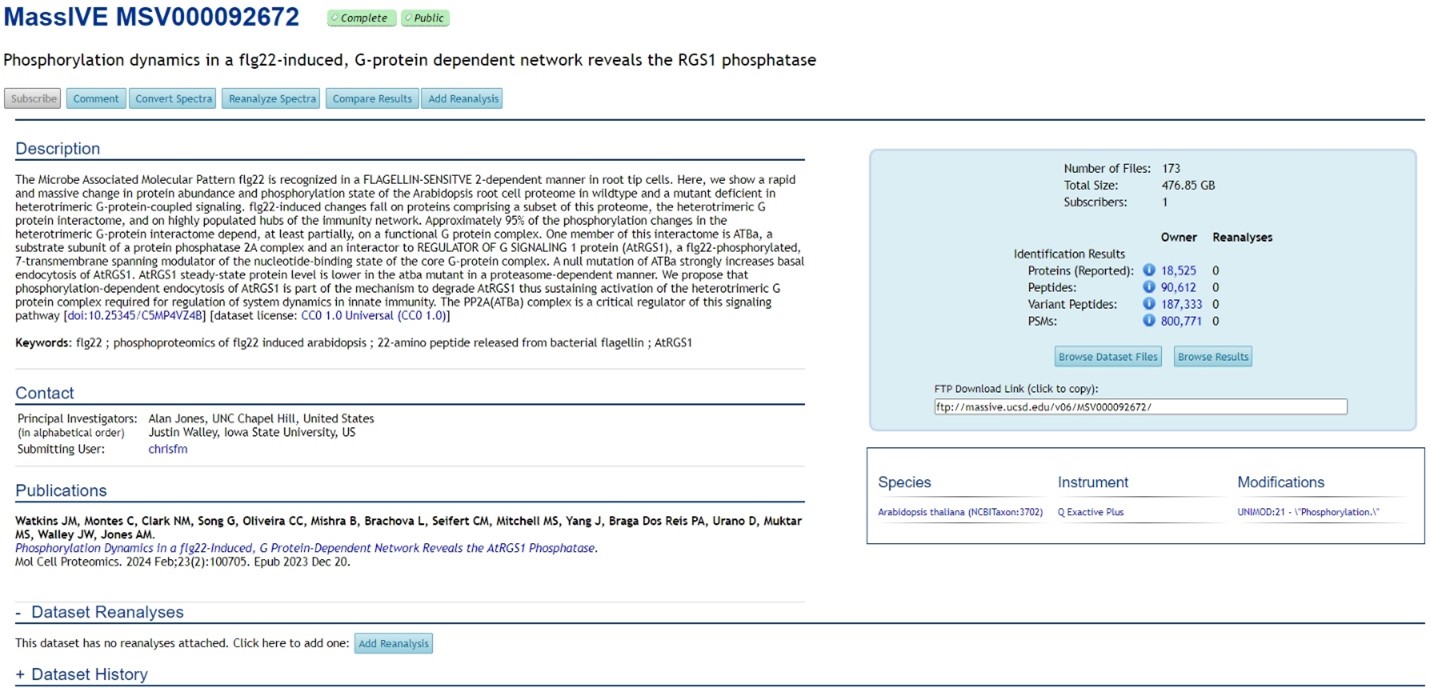
MassIVE captures metadata including description, keywords, contact information, species, instrument, modification, publication, dataset reanalysis, and dataset history. A unique feature of MassIVE is the reporting of the numbers of proteins, peptides, variant peptides, and Peptide-Spectrum Matches (PSMs) for identification datasets, as well as numbers of differentially expressed proteins and quantified proteins for quantification datasets.
The MassIVE search portal supports searches with peptide sequences (unmodified or modified), post-translational modifications, protein names, and accession numbers, and MassIVE can export search results containing metadata into a TSV file. Each spectrum in MassIVE is assigned a Universal Spectrum Identifier (USI), so that it can be searched at MassIVE’s USI portal.
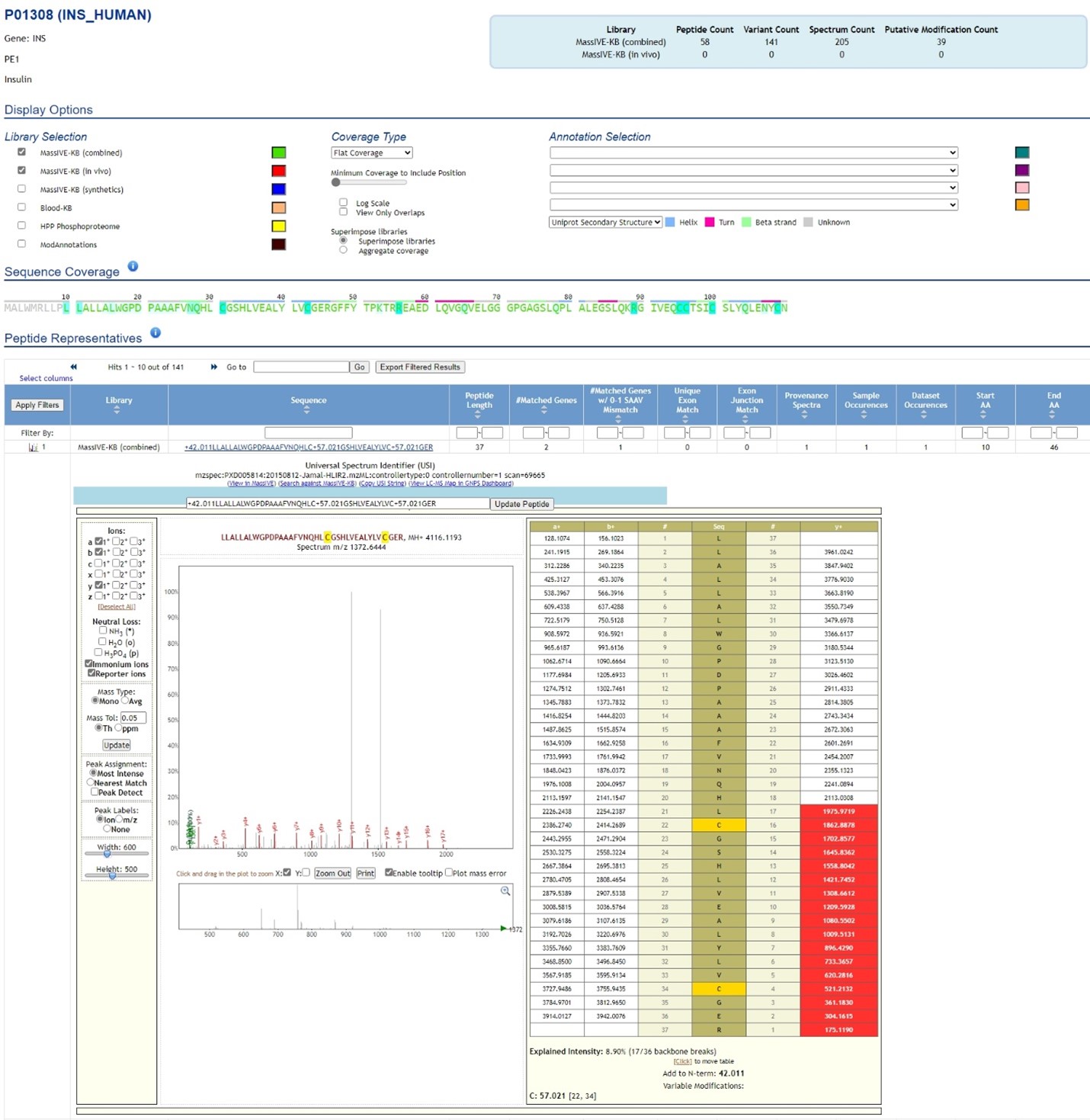
MassIVE’s Protein Explorer is a powerful search functionality for users to explore the translated evidence and sequence coverage of nearly every human protein. The data used in the search query was generated by systematic reanalysis of 31 terabytes of public data from >20,000 LC/MS runs and including over 1 million synthetic peptide spectra. Interactive exploration of protein evidence includes coverage maps, functional sites, and full provenance and dataset mapping of every identified peptide (Figure 2).
As mentioned above, MassIVE serves not only as a mass spectrometry data repository, but also as an online environment for data analysis. A user account is required to access the data analysis environment. Users can reanalyze public datasets with the ProteoSAFe webserver and submit reanalysis results from the ProteoSAFe data analysis workflow or their own offline workflow back to MassIVE. Reanalysis results are archived in containers with unique container IDs (“RMSV” followed by nine digits).
Currently, there are dozens of analysis workflows available at the ProteoSAFe webserver. For example, the “Results Comparison” workflow will compare identification results between datasets or against any re-analyses of public data. Venn diagrams are used to compare results at the level of protein, peptide and spectrum identifications; agreements, disagreements and unique identifications can be interactively inspected for assessment of quality of identifications.
There are also a few useful workflows for data sharing. The “Convert Vender Spectrum Files to Open Format” workflow converts vendor raw files (MS data) to the open mzML format. The “Convert TSV Files to mzTab” workflow converts common tab-separated formats (identifications data) to the open mzTab format. The “Convert Search Engine Result Files to mzTab” workflow and “Convert mzIdentML files to mzTab” workflow are useful for file conversion as well.
There are a few extensions of MassIVE that are worth mentioning as well. MassIVE.quant is a repository infrastructure and data resource for reproducible quantitative proteomics. The MassIVE Knowledge Base (MassIVE-KB) is a set of peptide spectral libraries. MassIVE KB v1 was distilled from 658 million human proteomics higher energy collisional dissociation (HCD) Orbitrap MS/MS spectrum data. MassIVE KB v2 was constructed from 1.25 billion spectra from in vivo human proteomics and synthetic peptide experiments, including spectra from multiple data types. Finally, a subset of MassIVE datasets related to SARS-CoV-2/COVID19 is grouped together as CoronaMassKB.
In summary, MassIVE is a good repository choice for advanced proteomics researchers.
- iProX
iProX was initially established in 2017 by the National Key Research Program of China as an integrated proteome resources center to support the Chinese Human Proteome Project. Currently, it is the third largest proteomics data repository hosting about 10% of the datasets in ProteomeCentral. Because many MassIVE datasets are from data re-analysis, the number of direct submissions is higher in iProX than MassIVE. Datasets in iProX are treated as projects and organized by Project IDs (“IPX” followed by six digits, followed by “000”). Most datasets are submitted to ProteomeCentral as well and assigned PX Accession Numbers (“PXD” followed by six digits).
Each project can contain one or more subprojects organized by Subproject ID. For example, dataset PXD058593 is assigned Project ID IPX0007799000, and contains three Subprojects IPX0007799001, IPX0007799002, and IPX0007799003. Subprojects are not assigned separate PX Accession Numbers. iProX supports spectrum search at its own USI portal. The advanced search function in iProX allows users to search protein ID, peptide sequences, as well as common information in metadata. Although most datasets archived in iProX are submitted by Chinese researchers, it does accept submissions from other countries, including the United States.
- jPOST
The Japan ProteOme Standard Repository/Database (jPOST) was lunched by the Japan Science and Technology Agency in 2015. It consists of a repository component (jPOSTrepo) and a database component (jPOSTdb). Currently, less than 6% of the datasets in ProteomeCentral come from jPOST. In addition to mass spectrometry-based proteomics data, jPOSTrepo also accepts gel electrophoresis-based proteomics data, and antibody-based proteomics data. Although all iPOSTrepo datasets are assigned with jPOST identifiers (“JPST” followed by six digits), only mass spectrometry-based proteomics datasets are associated with PX Accession Numbers (“PXD” followed by six digits). Initially jPOSTrepo was designed for researchers from Asia and Oceania, but it has received submissions from other countries as well.
The database component, jPOSTdb, contains re-analysis results with unified criteria for proteome data from jPOSTrepo. jPOSTdb also provides graphics showing the frequency of detected post-translational modifications, the co-occurrence of phosphorylation sites on a peptide and peptide sharing among protein isoforms. In addition, jPOST also provides a few computational tools (jPOST gadget), including a database for in vitro human kinome (the complete set of protein kinases encoded in the genome) profiling, and a targeted proteomics platform for in vitro proteome-assisted MRM for protein absolute quantification.
In summary, jPOSTrepo is a candidate repository for sharing MS proteomics data, as well as 2D-PAGE and 2D-DIGE proteomics data.
- Panorama Public
Skyline is a free open-source software developed at the University of Washington for targeted proteomics method creation and quantitative data analysis. Skyline supports major targeted acquisition methods, including selected reaction monitoring (SRM)/multiple reaction monitoring (MRM), parallel reaction monitoring (PRM), data independent acquisition (DIA/SWATH) and data dependent acquisition (DDA). A review of Skyline can be found at this link. Skyline also works for generalized small molecules, such as lipidomics, glycomics, and metabolomics.
Panorama Public serves as the repository for mass spectrometry data processed by Skyline. Panorama Public has a built-in data visualization capability to allow researchers to share their analysis process. Instead of accession numbers, Panorama Public issues DOIs to datasets. In addition, Panorama Public datasets also submitted to ProteomeCentral are assigned PX Accession Numbers (“PXD” followed by six digits). Currently, only about 1% of the datasets in ProteomeCentral come from Panorama Public, and less than 60% of the datasets in Panorama Public have PX Accession Numbers.
- PeptideAtlas/PASSEL
Established by the Institute for Systems Biology in Seattle in 2002, PeptideAtlas is a multi-organism, publicly accessible compendium of peptides identified in a large set of tandem mass spectrometry proteomics experiments. The long-term goal of the PeptideAtlas project is full annotation of genomes through a thorough validation of expressed proteins. The current “default” datasets in PeptideAtlas include seven human samples, three mouse samples, as well as samples from at least thirty-two other organisms. As a database, PeptideAtlas contains a wealth of searchable mass spectrometry information for proteins and peptides. As a repository, it archives the raw data used to build PeptideAtlas.
PeptideAtlas requires users to submit their data dependent acquisition (DDA) datasets to other PX Consortium repositories, and PeptideAtlas will pick up the dataset from there for reprocessing. PeptideAtlas also has several additional features. For example, PeptideAtlas was initially developed as an integral part of the Human Plasma Proteome Project (HPPP). Results of HPPP are hosted at Human Plasma Proteome Project Data Central at PeptideAtlas. In addition, PeptideAtlas has developed an automated system, Tiered Human Integrated Search Proteome, that integrates all major sources of human protein sequences into a set of searchable databases. PeptideAtlas also hosts the Spectrum Library Central as a resource for spectrum libraries, and enables spectrum search with SpectraST.
The SWATHAtlas is a resource for planning DIA/SWATH experiments, depositing published experimental high-quality spectral assay libraries, and exploring the results of uploaded datasets.
The SRMAtlas is a compendium of targeted proteomics assays to detect and quantify proteins in complex proteome digests by mass spectrometry and is intended as a resource for SRM/MRM-based proteomic workflows. The PeptideAtlas SRM Experiment Library (PASSEL) is a data repository that is designed to enable submission, dissemination, and reuse of SRM experimental results from analysis of biological samples. Currently, about 0.4% of datasets in ProteomeCentral are from PASSEL. Datasets in PASSEL are assigned accession numbers beginning with “PASS” followed by five digits. Most of these datasets are submitted to ProteomeCentral and receive PX Accession Numbers (“PXD” followed by six digits) as well. A unique feature of this resource is the PASSEL transition group browser, which allows users to inspect crucial parameters of SRM experiments (Figure 3).
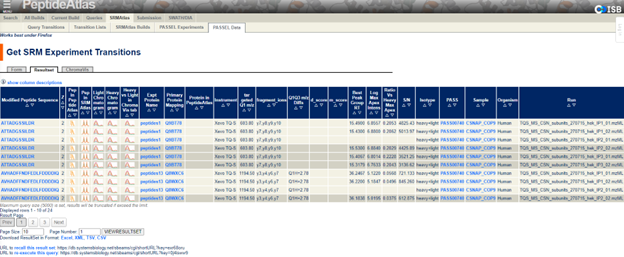
PASSEL is my recommendation to share SRM/MRM data, as PASSEL will process submitted raw data, and add high quality filtered results to SRMAtlas. In this way, researchers can directly contribute to the SRMAtlas whole-proteome catalogs of best available peptides and transitions to design SRM experiments.
This concludes my coverage of data repositories associated with the PX Consortium. In the last part of this blog series, I will introduce data repositories outside of the PX Consortium that are relevant to proteomics research.